
Pheochromocytoma: An Academic Review
1. Faiz Attari
2. Osmonova G. Zh.
(1. Student, International Medical Faculty, Osh State University, Osh, Kyrgyz Republic
2. Teacher, Dept. Of Pediatrics, International Medical Faculty, Osh State University, Osh, Kyrgyz Republic.)
Abstract
Pheochromocytoma are rare neuroendocrine cancers that arise from chromaffin cells within the adrenal medulla, because of their excessive secretion of catecholamines. Although these are rare tumors, they can be quite challenging to diagnose and manage because they can present for severe cardiovascular complications. It can often occur in association with hereditary syndromes such as Multiple Endocrine Neoplasia type 2 (MEN2), Von Hippel-Lindau disease (V Neurofibromatosis type 1 (NF1). This report contains an academic overview of pheochromocytoma, formatted following the IMRAD pattern—Introduction, Methods, Results, and Discussion, with the exceptions that pertain to pathogenesis. The paper integrates the findings from recent scientific studies regarding its epidemiology, presentation, means of diagnosis, treatment modalities, and prognosis. The paper also ends with an assessment concerning recent advancements in the means of diagnosis and treatment.
Introduction
Pheochromocytoma is defined as an endocrine tumor that produces catecholamines from chromaffin cells that are located in or near the adrenal medulla. The cells produce and secrete an excess of epinephrine, norepinephrine, and dopamine into the bloodstream in amounts that are in excess and result in systemic effects that cause episodic or persistent hypertension. Pheochromocytoma malignant behaviour, described by local invasiveness or metastasis.
Epidemiology
"Pheochromocytoma" is defined as that endocrine tumor that secretes catecholamines from chromaffin cells that are found in or close to the adrenal medulla. This tumour produces and secretes excessive amounts of epinephrine, norepinephrine, and dopamine into the general circulation in amounts that are in excess and cause systemic effects that produce episodic or sustained hypertension.

Clinical Significance
Because it can result in potentially fatal cardiovascular crises, pheochromocytoma is clinically significant. Excessive catecholamine secretion causes tachycardia, hypertension, and metabolic problems. Preventing morbidity and mortality requires early detection and proper treatment. Improvements in imaging, biochemical testing, and surgical methods have enhanced patient outcomes and diagnostic precision.
Methods
Literature Search Strategy
Using databases like PubMed, Scopus, and Web of Science, a systematic review of the literature was carried out. "Pheochromocytoma," "adrenal tumor," "catecholamine-secreting tumor," "diagnosis," "management," and "genetic syndromes" were the search terms utilized. To ensure current relevance, articles published between 2000 and 2024 were included.
Inclusion and Exclusion Criteria
Peer-reviewed clinical studies, meta-analyses, and reviews with a human subject focus were all included in the inclusion criteria. Priority was given to studies that addressed clinical characteristics, diagnostic techniques, and management approaches. Animal studies, case reports with no clinical significance, and articles not available in English were among the exclusion criteria.
Data Extraction and Analysis
Epidemiology, clinical presentation, diagnostic techniques, treatment results, and genetic correlations were among the data that were extracted. To find patterns and consensus in the literature, quantitative data were tabulated and qualitative findings were combined.
Results
Clinical Presentation
Catecholamine excess is the main cause of pheochromocytoma's clinical symptoms. Sweating, palpitations, and intermittent headaches make up the classic triad. Presentations, however, differ greatly, and some patients do not exhibit any symptoms until they are unintentionally discovered during imaging for unrelated conditions.
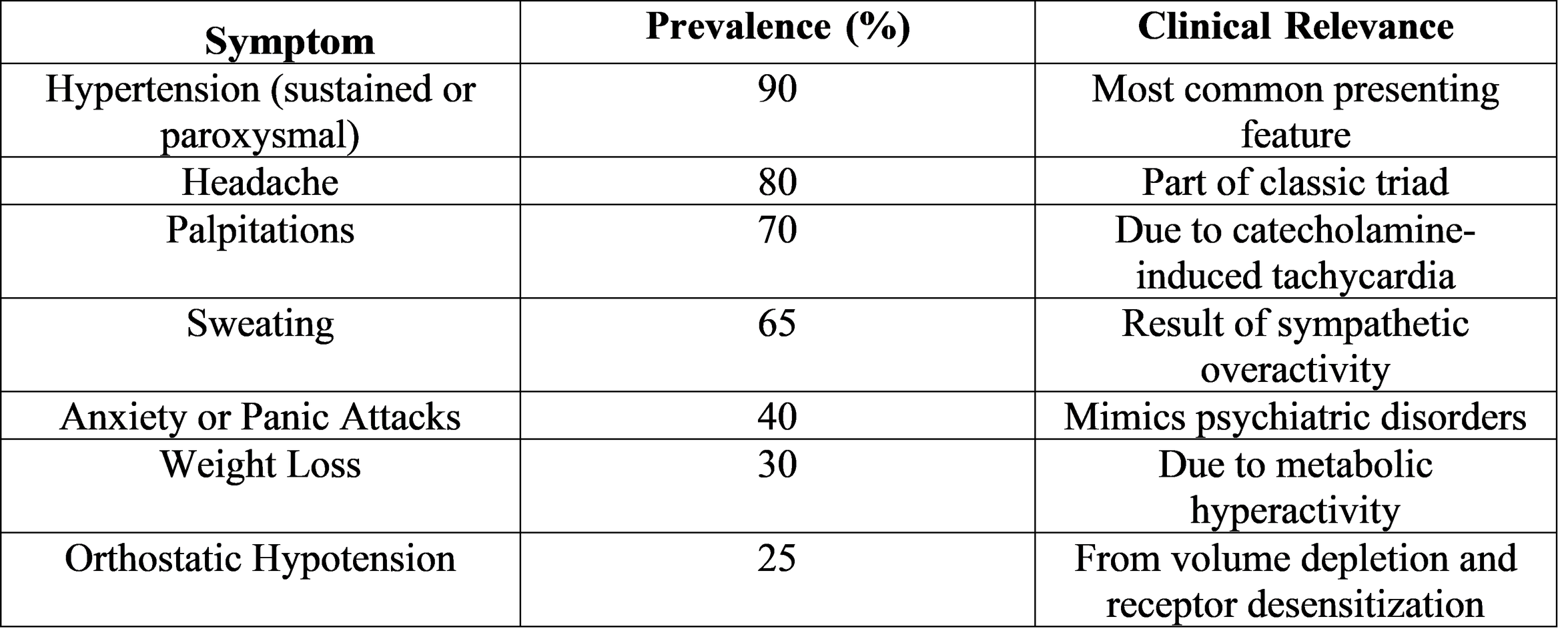
Biochemical Diagnosis
The mainstay of diagnosis is still biochemical testing. The highest sensitivity and specificity can be obtained by measuring urinary fractionated metanephrines or plasma free metanephrines.

Medication, stress, or inadequate sampling conditions can all lead to false positives. Clinical correlation and confirmatory testing are therefore crucial.
Imaging Modalities
Imaging is used to locate the tumor after biochemical confirmation is obtained. While functional imaging helps identify multifocal or metastatic disease, computed tomography (CT) and magnetic resonance imaging (MRI) are first-line modalities.
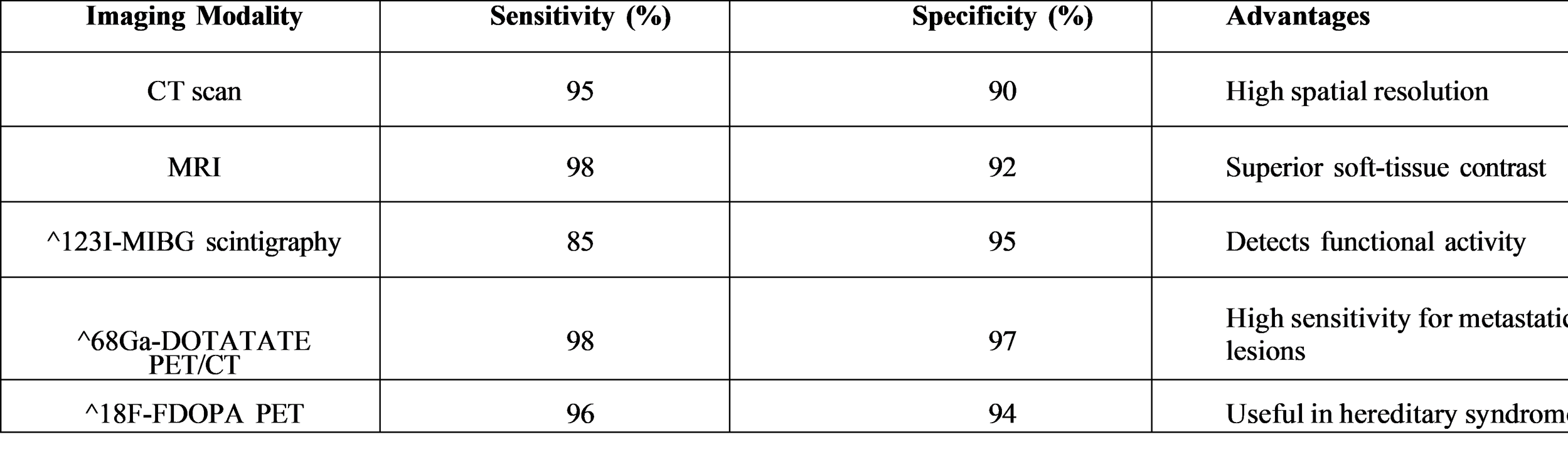
Genetic Associations
Mutations in a number of susceptibility genes are found through genetic testing. RET (MEN2), VHL, NF1, and SDHx gene mutations are the most prevalent. All patients should receive genetic counseling because hereditary forms affect surveillance and treatment plans.

Management Strategies
The definitive treatment for pheochromocytoma is surgical resection. Preoperative medical optimization is critical to prevent intraoperative hypertensive crises.
Preoperative Management
Alpha-adrenergic blockade starts 10 to 14 days before surgery to manage blood pressure and increase plasma volume. Doctors often use phenoxybenzamine or doxazosin. They add beta-blockers only after achieving sufficient alpha blockade to manage tachycardia.
Surgical Approaches
Laparoscopic adrenalectomy is the preferred approach for localized tumors due to reduced morbidity and faster recovery. Open surgery is reserved for large or invasive tumors.

Postoperative Care and Follow-Up
Postoperative monitoring includes blood pressure assessment and biochemical testing to confirm complete tumor removal. Lifelong follow-up is recommended due to the risk of recurrence or metastasis, particularly in hereditary cases.
Discussion
Diagnostic Challenges
The challenge in diagnosing pheochromocytoma has remained owing to its non-specificity and its overlap in symptoms with that of other ailments, such as the anxiety disorder and the essential hypertension.
With the advancement in biochemical tests, the misinterpretation or false-positive results also form a challenge.
Genetic Insights and Clinical Implications
Genetic mutations Identification has transformed the understanding of pheochromocytoma. Genetic analysis not only provides assistance in reaching the diagnosis but also in monitoring for associated neoplasias. For example, in RET mutations, there is screening for medullary thyroid carcinoma, and in VHL mutations, there is screening for renal carcinoma.
Surgical and Medical Advances
Minimally invasive surgical techniques have largely reduced perioperative morbidity. It is still essential to give preoperative alpha blockade, which is the main measure that avoids
hypertensive crises during tumor manipulation. New pharmacologic agents, for example, selective alpha, 1 blockers and calcium channel blockers, become the better options for hemodynamic control.
Prognosis and Outcomes
After a total resection, the survival rate of patients with benign pheochromocytoma is very good, being over 90% at 10 years. Thus, the main threat of these tumors comes from the rare ones that undergo malignant transformation and have a less favorable prognosis: 5, year survival rates vary between 40 and 60%. Some of the factors that influence the outcome are the size of the tumor, genetic background, and whether metastases are present.

Emerging Research and Future Directions
Most of the recent studies revolve around molecular imaging, targeted therapies, and immunotherapy. DOTATATE PET/CT has been largely accepted as a leading imaging technique for the identification of metastatic disease. There are a number of new drug candidates targeting angiogenesis and metabolic pathways that have not yet been approved but are being researched. On top of that, AI and machine learning technologies are also considered for use in diagnostic prediction and risk stratification.
Conclusion
Pheochromocytoma continues to be a complicated and highly important clinically challenging endocrine tumor that demands the involvement of various medical specialties for its best management. Improvements in biochemical tests, imaging, and genetics have greatly changed the accuracy of diagnosis and the results of patients. It is worth noting that the timely diagnosis and correct preoperative preparation are the main factors in avoiding the occurrence of potentially fatal complications. The removal of the tumor through surgery is still the primary option, with the use of minimally invasive methods giving great results in cases where the disease is confined to one area.
One of the major factors genetic testing is able to do is to identify hereditary syndromes, help surveillance and give family counseling information. Even with these innovations, the problems continue to exist in the identification of cancerous and recurrent cases. Research directions should be aimed at improving molecular diagnostics, inventing targeted therapies, and using artificial intelligence in clinical decision, making.
Moreover, pheochromocytoma is a prime example of the convergence of endocrinology, genetics, and surgical innovation. The effect of continuous research and teaming up with different disciplines will be to deepen the knowledge, advance the patient care, and lessen the disease and death rate caused by this infrequent but powerful neoplasm.
References
Eisenhofer, Graeme, et al. “Biochemical Diagnosis of Pheochromocytoma: How to Distinguish True- from False-Positive Test Results.” Journal of Clinical Endocrinology & Metabolism, vol. 103, no. 1, 2018, pp. 1–12.
Fishbein, Lauren, et al. “Pheochromocytoma and Paraganglioma: Genetics, Diagnosis, and Treatment.” Endocrine Reviews, vol. 38, no. 6, 2017, pp. 489–515.
Lenders, Jacques W. M., et al. “Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline.” Journal of Clinical Endocrinology & Metabolism, vol. 99, no. 6, 2014, pp. 1915–1942.
Neumann, Hans-Peter H., et al. “Genetic Basis of Pheochromocytoma and Paraganglioma.” New England Journal of Medicine, vol. 381, no. 6, 2019, pp. 552–565.
Pacak, Karel, et al. “Recent Advances in the Genetics, Diagnosis, and Treatment of
Pheochromocytoma and Paraganglioma.” New England Journal of Medicine, vol. 379, no. 7, 2018, pp. 593–600.
Taïeb, David, et al. “Current Approaches and Recent Developments in the Management of Pheochromocytoma and Paraganglioma.” Endocrine Reviews, vol. 41, no. 2, 2020, pp. 1–35.
Zelinka, Tomas, et al. “High Incidence of Cardiovascular Complications in Patients with
Pheochromocytoma.” Journal of Hypertension, vol. 30, no. 11, 2012, pp. 2044–2050.
Young, William F. “Pheochromocytoma and Paraganglioma in Adults: Clinical Presentation and Diagnostic Evaluation.” UpToDate, 2023.