
Hereditary Hemolytic Anemia in Children
1. Endesh kyzy Gulsara
2. Amannum Arah
(1. Teacher, International Medical Faculty, Osh State University, Osh, Kyrgyz Republic.
2. Student, International Medical Faculty, Osh State University, Osh, Kyrgyz Republic.)
Abstract
Hereditary hemolytic anemia (HHA) represents a heterogeneous group of inherited disorders characterized by premature destruction of red blood cells (RBCs) due to intrinsic defects. These defects involve abnormalities in RBC membrane structure, hemoglobin synthesis, or enzymatic pathways. HHA is a significant cause of chronic anemia in pediatric populations worldwide. Clinical manifestations range from asymptomatic compensated hemolysis to severe anemia requiring transfusion. Advances in diagnostic technologies, including molecular testing, have improved early detection and classification. This article reviews the epidemiology, pathophysiology, clinical presentation, diagnostic approach, complications, and management strategies of hereditary hemolytic anemia in children.
Hereditary hemolytic anemias (HHAs) constitute a diverse group of genetic disorders characterized by the premature destruction of erythrocytes. In the pediatric population, these conditions present significant diagnostic and therapeutic challenges due to their varied clinical manifestations, ranging from asymptomatic mild anemia to life-threatening crisis and transfusion dependency.
Keywords
Hereditary hemolytic anemia, pediatric anemia, membranopathy, enzymopathy, hemoglobinopathy, hemolysis, thalassemia, G6PD deficiency
Introduction
Hemolytic anemia occurs when RBC destruction exceeds production, resulting in reduced circulating red cell mass.
Normal RBC lifespan = ~120 days, but in hemolysis → few days to weeks.
Children are particularly vulnerable due to:
-Immature immune system
-Higher metabolic demands
-Genetic predisposition (e.g., hemoglobinopathies)
Hereditary hemolytic anemia (HHA) is defined as a group of inherited disorders in which red blood cells are destroyed prematurely due to intrinsic cellular defects. Unlike acquired hemolytic anemia, these conditions arise from genetic mutations affecting RBC structure or function.
In children, HHA contributes significantly to chronic anemia and morbidity. Early diagnosis is crucial to prevent complications such as growth retardation, organ damage, and transfusion-related issues.
Hereditary hemolytic anemia (HHA) is a group of genetic disorders characterized by the premature destruction of red blood cells (RBCs). While a normal red blood cell lives for about 120 days, in children with HHA, the lifespan of these cells is significantly shortened. When the bone marrow cannot produce new cells fast enough to replace those being destroyed, anemia occurs.
Epidemiology
Hereditary hemolytic anemias (HHAs) are a diverse group of genetic disorders characterized by the premature destruction of red blood cells (RBCs). In a healthy child, RBCs typically circulate for about 120 days; however, in HHA, the lifespan is significantly shortened. Because these conditions are inherited, they often present in early infancy or childhood, requiring lifelong management.
AIHA incidence: 1–3 cases per 100,000 children/year
Common causes in hospitalized children:
Thalassemia major (~27%)
Hereditary spherocytosis
G6PD deficiency
Classification and Types of HHA
The causes of hereditary hemolysis are generally categorized by which part of the red blood cell is structurally or functionally defective.
1. Erythrocyte Membrane Defects
These disorders occur due to mutations in proteins that provide the RBC with its skeletal integrity and flexibility.
Hereditary Spherocytosis (HS): The most common membrane defect. It results in sphere-shaped cells that are easily trapped and destroyed by the spleen.
Hereditary Elliptocytosis (HE): Characterized by cigar-shaped RBCs. While often asymptomatic, some pediatric cases can present with severe hemolysis.
2. Red Blood Cell Enzymopathies
Enzymes are required to protect RBCs from oxidative stress and to generate energy.
Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency: The most prevalent enzyme disorder. Hemolysis is often episodic, triggered by infections, specific foods (fava beans), or certain medications.
Pyruvate Kinase (PK) Deficiency: A less common but often more severe chronic hemolytic anemia involving the glycolytic pathway.
3. Hemoglobinopathies
These involve genetic defects in the globin chains of the hemoglobin molecule.
Sickle Cell Disease (SCD): Caused by a point mutation leading to Hemoglobin S. Under low oxygen, cells "sickle," causing both hemolysis and painful vascular blockages.
Thalassemias: Quantitative defects where the body produces insufficient alpha or beta globin chains, leading to ineffective erythropoiesis and hemolysis.
Pathophysiology
In a healthy child, red blood cells are flexible, biconcave discs that easily squeeze through tiny capillaries. In HHA, a genetic "glitch" affects one of three main components of the cell, leading to its destruction (hemolysis):
The Membrane: Defects in the cell's "skin" make it fragile (e.g., Hereditary Spherocytosis).
The Enzymes: Deficiencies in energy-producing enzymes prevent the cell from protecting itself against oxidative stress (e.g., G6PD Deficiency).
The Hemoglobin: Mutations in the oxygen-carrying protein cause the cell to become rigid or misshapen (e.g., Sickle Cell Disease or Thalassemia).
Pediatric Impact
In children, the epidemiological burden is significant. In "malaria-endemic" belts, HHA is a leading cause of childhood morbidity. However, due to global migration, these conditions are now frequently diagnosed in pediatric clinics worldwide, regardless of the local climate.
Note: Early screening (such as Newborn Screening programs) has significantly reduced mortality rates for Sickle Cell Disease and Thalassemia in many developed nations.
Clinical Presentation
Children with HHA often present with a "classic triad" of symptoms. Because the destruction of RBCs releases bilirubin (a yellow pigment) into the bloodstream, the clinical signs are often quite visible:
Anemia: Paleness, fatigue, and shortness of breath during play.
Jaundice: Yellowing of the eyes (sclera) and skin.
Splenomegaly: An enlarged spleen, as it works overtime to filter out the damaged blood cells.
Recent research on haemolytic anemia (HA) in children highlights a shift in understanding the prevalence, underlying causes, and the impact of recent global events (like COVID-19) on diagnosis rates. Hemolytic anemia occurs when red blood cells are destroyed faster than the bone marrow can produce them, leading to a shortage of oxygen-carrying cells.
Key Research Findings
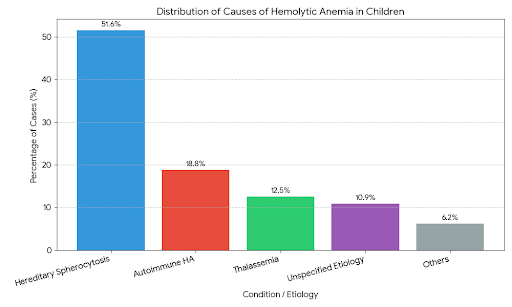
Dominance of Inherited Causes: Studies, such as those conducted in Eastern Europe and Central Asia, indicate that Hereditary Spherocytosis remains the most common cause of pediatric haemolytic anemia, often accounting for over 50% of new diagnoses. This is followed by autoimmune types and thalassemias.
Autoimmune Hemolytic Anemia (AIHA) Complexity: Recent research (2024) published in Blood and MDPI shows that pediatric AIHA is frequently associated with underlying immune disorders. Approximately 30% of cases are secondary to conditions like primary immunodeficiency or Evans Syndrome. These cases often require more aggressive, tailored treatments (e.g., Rituximab or IVIG) compared to idiopathic cases.
Post-COVID-19 Trends: A 2024 cross-sectional study noted a significant increase in the incidence of haemolytic anaemia admissions in emergency departments during and after the COVID-19 pandemic period (2020–2023), suggesting a potential link between viral triggers or immune dysregulation and hemolytic episodes.
Global Prevalence: While general anemia affects nearly 40% of children aged 6 months to 5 years globally, haemolytic anemia is rarer (AIHA affects roughly 1 in 80,000 children) but carries higher risks of complications like gallstones, kidney failure, and heart failure if not managed early.
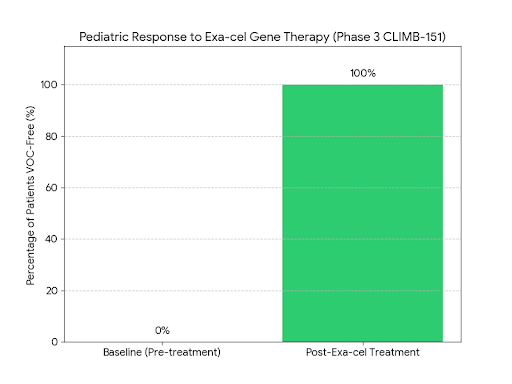
1. Breakthrough in Gene Therapy (Exa-cel)
In 2024–2025, the FDA and global health bodies approved Exagamglogene autotemcel (Exa-cel) for children as young as 12, with trials for children aged 5–11 showing massive success.
Finding: This CRISPR-based therapy "turns back on" fetal hemoglobin, effectively curing Sickle Cell Disease and Beta-Thalassemia.
Impact: In the Phase 3 CLIMB-151 study (2025), 100% of evaluable pediatric patients were free from painful vaso-occlusive crises for over a year.
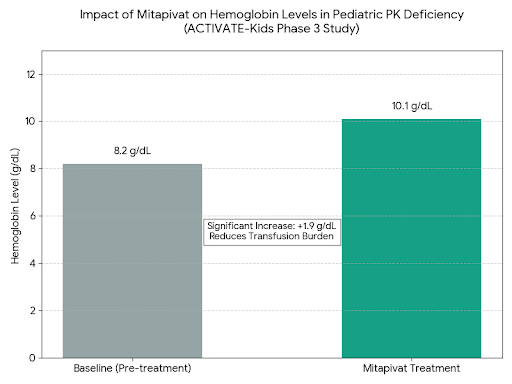
Research Breakthrough: Mitapivat for Pediatric PK Deficiency
The Finding: A Significant Hemoglobin Boost
The global Phase 3 ACTIVATE-Kids study focused specifically on children who were not dependent on regular blood transfusions. The core finding was that Mitapivat, an oral small-molecule activator of the PK enzyme, successfully "re-energized" the red blood cells.
Hemoglobin Response: Evaluated patients showed a sustained increase in hemoglobin levels, with a mean increase of approximately +1.9 g/dL from their baseline.
Target Achievement: This increase was achieved using a daily oral pill, which is far less invasive than the historical standard of care.
The Impact: Moving Away from Transfusions
For decades, children with severe PK deficiency had few options besides frequent, painful blood transfusions and splenectomy (removal of the spleen).
Disease-Modifying Therapy: Mitapivat is the first-ever oral therapy that addresses the underlying cause of the disease rather than just treating the symptoms.
Transfusion Reduction: By stabilizing red blood cells and raising natural hemoglobin levels, the drug significantly reduces the "transfusion burden," lowering the risk of iron overload and organ damage that often complicates long-term transfusion therapy in children.
Visualization of Clinical Results
The graph below illustrates the average increase in Hemoglobin (Hgb) levels observed in pediatric patients during the Phase 3 trial, demonstrating the shift from a state of moderate anemia (baseline) to a more stable physiological level.
This advancement is currently paving the way for Mitapivat to become the new global standard of care for children as young as 1 year old suffering from this rare genetic anemia.
Conclusion
Hemolytic anemia in children is a complex disorder with diverse etiologies and clinical presentations. Early recognition, appropriate laboratory evaluation, and targeted therapy are essential for optimal outcomes. Advances in immunotherapy and supportive care have significantly improved prognosis, but challenges remain in diagnosis and management, particularly in resource-limited settings.
References
1.Children’s National Hospital. Hemolytic anemia in children.
2.Arora S, et al. Autoimmune hemolytic anemia in children.
3.Blackall D, et al. AIHA in children: review.
4.Malik H, et al. Hemolytic disease of fetus and newborn.
5.BMJ Paediatrics Open. Hemolytic anemia in children study.
6.StatPearls. Hemolytic anemia complications