
Acquired Hemolytic Anemia in Children
1. Abhishek Raja
2. Osmonova G. Zh.
(1. Student, International Medical Faculty, Osh State University, Osh, Kyrgyz Republic.
2. Teacher, International Medical Faculty, Osh State University, Osh, Kyrgyz Republic.)
Abstract
Acquired hemolytic anemia in children encompasses a heterogeneous group of disorders characterized by the premature destruction of RBCs due to extrinsic factors acting on otherwise structurally normal erythrocytes. The condition ranges from mild, self-limited hemolysis to fulminant, life-threatening anemia. Immune-mediated mechanisms, in particular, autoimmune hemolytic anemia, prevail in the pediatric age group, whereas the nonimmune causes-to wit, infections, drugs, mechanical injury, and systemic diseases-play a contributory role to a very considerable extent. This enhanced review encompasses epidemiology, immunopathogenesis, molecular mechanisms, detailed clinical correlations, advanced diagnostics, management algorithms, complications, prognosis, and preventive strategies, and proposed graphs and diagrams for academic use.
Introduction and Definitions
Hemolytic Anemia is a type of anemia caused because of decreased red blood cell survival rates (<120 days). This is due to the rate of its destruction exceeding the bone marrow’s compensatory capacity. There are several types of hemolytic anemias among children. They can be categorized as follows
· Inherited (intrinsic) hemolytic anemias – membrane defects, enzyme deficiencies, hem
· Acquired or extrinsic anemias – immune and nonimmune destruction of normal erythrocytes
Sickle cell hemolytic anemia is a significant acquired hemolytic anemia because of its acute onset, potential reversibility, and association with systemic disease. Early diagnosis is important, especially in infants and young children, whose acute hemoglobin loss can lead to cardiac failure.
Epidemiology
Acquired hemolytic anaemia is less common in children than inherited forms but disproportionately accounts for many hematologic emergencies.
· Incidence of pediatric AIHA: ~0.2–0.4 cases per 100,000 children per year
· Bimodal age distribution: infancy (<2 years) and adolescence
· Slight female predominance, particularly in autoimmune‑associated cases
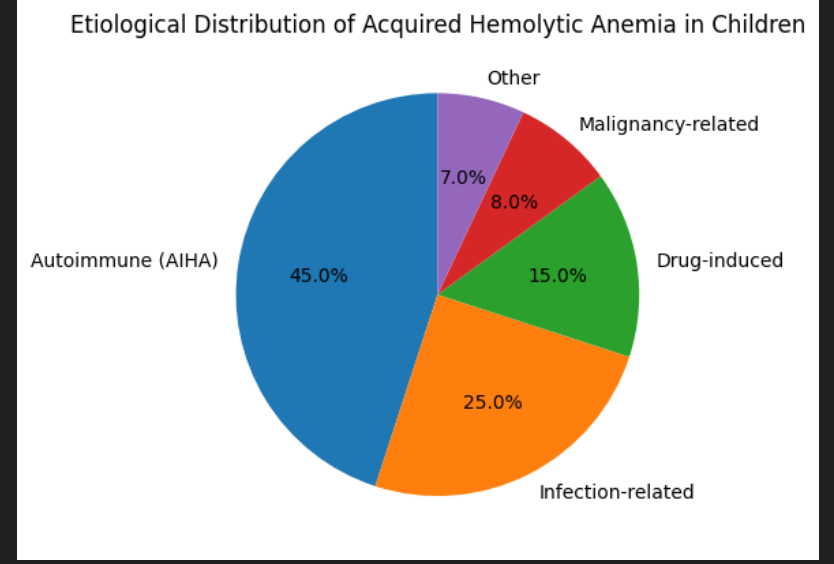
· Secondary AIHA accounts for up to 40–50% of cases in tertiary‑care cohorts
Thus, geographical variation exists due to differences in infectious disease prevalence, access to vaccination, and diagnostic capacity.
Classification of Acquired Hemolytic Anemia
3.1 Immune-mediated Hem
3.1.1. Autoimmune Hem
AIHA occurs when autoantibodies target RBC antigens on the surface of red cells, resulting in their premature destruction.
The classification based on thermal reactivity:
· Warm AIHA (IgG, 37°C active
· Cold agglutinin disease (IgM, active
· Mixed AIHA
· Paroxysmal cold hemoglobinuria: This
Based on Etiology:
· Primary (idiopathic)
· Secondary (Infection-linked, Autoimmune disease, malignancy
3.1.2 Alloimmune Haemolytic Anaemia
Post‑transfusion hemolysis
Haemolytic disease due to maternal antibodies (rare beyond neonatal period)
3.2 Non‑Immune Acquired Haemolytic Anaemia
Infection‑related hemolysis
Drug‑induced non‑immune hemolysis
Microangiopathic hemolytic anemia (MAHA)
Mechanical hemolysis
Hypersplenism
Immunopathogenesis
4.1 Mechanisms of Autoantibody
Autoantibody production can be triggered by the
Molecular mimicry following infection
Immune tolerance loss
Polyclonal B-cell Activation
Dysfunction of regulatory T cells
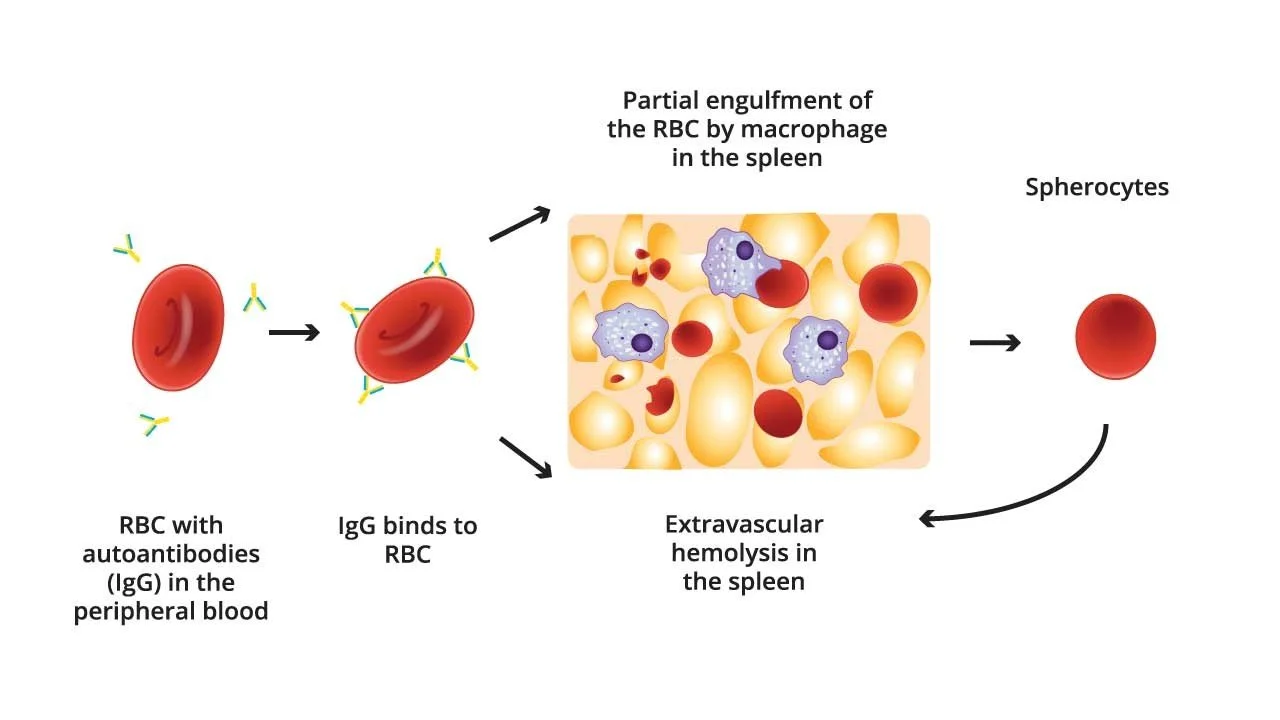
4.2 Ex- & In vivo Hem
· Hemolysis outside
· IgG-coated RBCs identified by Fc receptors of splenic macrophages
· Partial Phagocytosis → Spherocyte
· Most common in Warm AIHA
· Intravascular Hem
· Complement activation (C5-C9)
· Release of free hemoglobin in plasma
· Identified in cold antibody disease and PCH
Infection‑Associated Acquired Hemolysis
Common triggers include:
Viral: EBV, CMV, influenza, hepatitis viruses
Bacterial: Mycoplasma pneumoniae, Streptococcus pneumoniae
Parasitic: Malaria (non‑immune and immune mechanisms)
Mechanisms include:
RBC invasion, direct
Immune complex formation
Complement activation
c Cytokine-mediated membrane damage
Drug‑Induced Hemolytic Anemia
6.1 Immune Drug-Induced hemolysis
Hapten-induced (Penicillin type )
Immune complex type (Quinidine)
Autoantibody induction (methldopa)
6.2 Non-Immune Drug induced hemolysis
Oxidative stress (particularly in G6PD deficiency)
Direct membrane toxicity
Common implicated drugs:
Antibiotics (cephalos
Antimal
Antiepilept
Clinical Manifestations
7.1 General Symptoms
Pallor
Fatigue
Dyspnea
Poor feeding (infants)
7.2 Hemolysis‑Specific Features
Jaundice
Dark or cola‑colored urine
Splenomegaly
Gallstone‑related abdominal pain (chronic hemolysis)
7.3 Severe Presentations
Acute hemolytic crisis
High‑output cardiac failure
Shock (rare but possible)
Diagnostic Evaluation
8.1 Laboratory Markers of Hemolysis
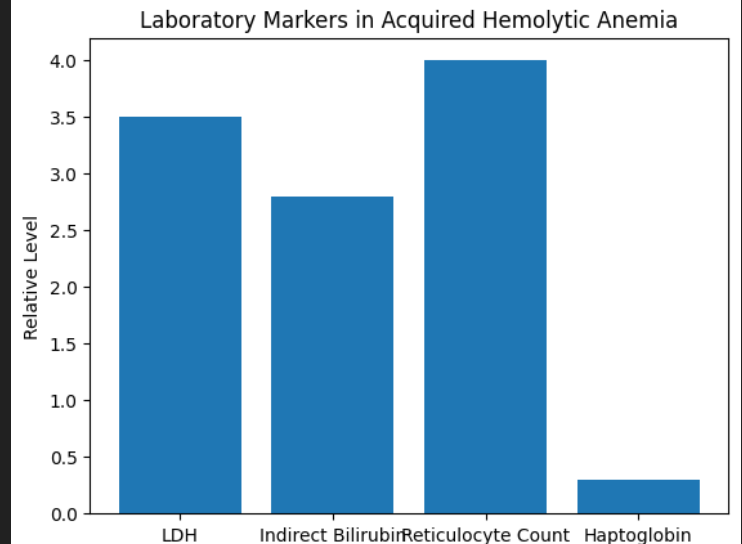
Reticulocytosis (unless marrow suppressed)
Elevated indirect bilirubin
Increased LDH
Reduced haptoglobin
8.2 Immunohematologic Tests
Direct antiglobulin test (DAT)
IgG positive
C3d positive
Indirect antiglobulin test (selected cases)
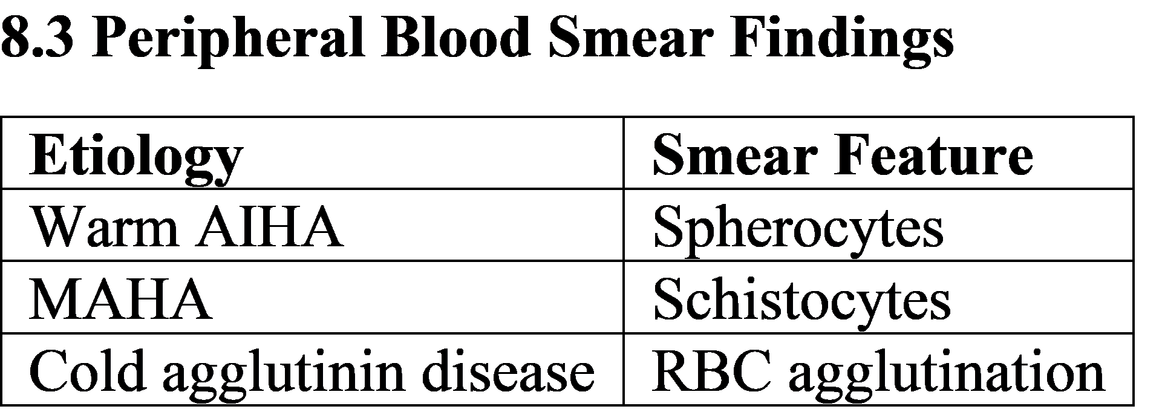
Management Strategies
10.1 Emergency Management
Stabilization (ABC)
Oxygen therapy
Packed RBC transfusion (cross‑match challenges anticipated)
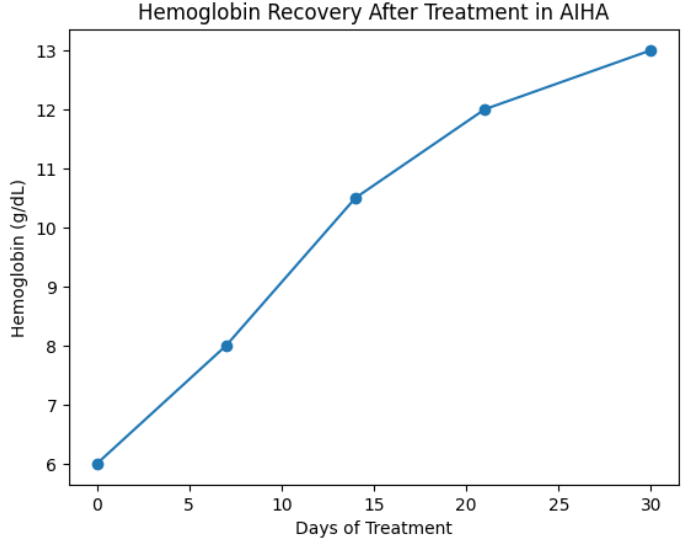
10.2 Pharmacologic Therapy
First‑line:
Corticosteroids (prednisolone 2–4 mg/kg/day)
Second‑line:
IVIG
Rituximab
Third‑line / Refractory:
Immunosuppressants
Splenectomy (carefully selected cases)
Special Situations
11.1 Acquired Hemolytic Anemia in Infancy
Often post‑viral
Higher risk of rapid decompensation
Usually good response to steroids
11.2 Hemolysis in Autoimmune Diseases
SLE‑associated AIHA
Evans syndrome (AIHA + ITP)
Complications
Pigment gallstones
Chronic anemia
Iron overload (repeated transfusions)
Thromboembolism
Infections due to immunosuppression
Prognosis and Outcomes
Primary AIHA: good prognosis, high remission rates
Secondary AIHA: relapse common
Mortality is low with modern therapy but increases with delayed diagnosis
Prevention and Follow‑Up
Vaccination against encapsulated organisms
Infection prevention
Long‑term monitoring for relapse
Conclusion
Acquired hemolytic anemia in children is a multifaceted disorder requiring a thorough understanding of immunologic mechanisms, vigilant diagnostic evaluation, and individualized therapy. Advances in immunomodulatory treatment have significantly improved outcomes. Early recognition and multidisciplinary care remain the cornerstone of successful management.
REFERENCES
Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM.
Nelson Textbook of Pediatrics. 21st ed. Philadelphia: Elsevier; 2020.
Chapter: Hemolytic Anemias in Children.Orkin SH, Fisher DE, Ginsburg D, Look AT, Lux SE, Nathan DG. Nathan and Oski’s Hematology and Oncology of Infancy and Childhood. 8th ed. Philadelphia: Elsevier; 2015.
Hoffbrand AV, Higgs DR, Keeling DM, Mehta AB. Postgraduate Hematology. 7th ed. Wiley-Blackwell; 2016.
Greer JP, Arber DA, Glader B, et al. Wintrobe’s Clinical Hematology. 14th ed. Wolters Kluwer; 2019.
Hill QA, Stamps R, Massey E, Grainger JD, Provan D, Hill A. The diagnosis and management of primary autoimmune hemolytic anemia. British Journal of Haematology. 2017;176(3):395–411.
Michel M. Autoimmune hemolytic anemia. Hematology Am Soc Hematol Educ Program. 2015;2015(1):387–396.
Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Hemolytic anemias in children.
The Lancet. 2017;390(10091):311–323.Sokol RJ, Booker DJ, Stamps R. Autoimmune hemolysis: an 18-year study of 865 cases referred to a regional transfusion center. British Medical Journal. 1992;305:156–159.
Rothman JA, Weitz IC. Autoimmune hemolytic anemia in children.
Pediatric Clinics of North America. 2013;60(6):1489–1511.Teachey DT, Manno CS. Autoimmune cytopenias in childhood. Seminars in Hematology. 2005;42(4):299–310.
Barcellini W, Fattizzo B. Clinical applications of hemolytic markers in autoimmune hemolytic anemia.
Disease Markers. 2015;2015:635670.