
Pediatric Gonadal Diseases
1. Gulnaz Osmonova
2. Swati Bhupesh
(1. Teacher, International Medical Faculty, Osh State University, Osh, Kyrgyz Republic.
2. Student, International Medical Faculty, Osh State University, Osh, Kyrgyz Republic.)
Abstract
Pediatric gonadal diseases encompass a heterogeneous group of congenital and acquired conditions affecting the testes and ovaries that present across neonatal, childhood and adolescent periods. These include disorders/differences of sex development (DSD), undescended testis (cryptorchidism), gonadal tumours (germ cell and sex cord-stromal tumours), testicular torsion, autoimmune or genetic gonadal failure, gonadal dysgenesis, and traumatic or infectious injuries. Advances in molecular genetics, imaging, and multidisciplinary care have significantly changed diagnostic algorithms and management strategies. Early identification, timely surgical or medical intervention, careful fertility preservation consideration, and long-term endocrine and psychosocial follow-up are central to improving outcomes. This review summarizes current concepts in epidemiology, pathophysiology, clinical presentation, diagnostic evaluation (including first-line biochemical tests and modern genetic testing), and evidence-based management approaches for major pediatric gonadal conditions. We highlight contemporary consensus statements and guidelines that emphasize multidisciplinary teams, individualized treatment planning, shared decision-making with families, and the need for life-long follow-up in many conditions. Practical algorithms for initial triage, suggested timelines for interventions (e.g., timing of orchidopexy), principles of gonadal tumour management, and fertility preservation strategies are presented. Finally, we identify gaps in evidence and propose priorities for research and registry-based surveillance.
Keywords: pediatric gonadal disease, disorders/differences of sex development, cryptorchidism, gonadal tumors, testicular torsion, fertility preservation, pediatric endocrinology
Introduction
Gonadal diseases in children and adolescents present a unique intersection of endocrine, surgical, oncologic, genetic, and psychosocial care. The gonads have roles in sex differentiation, pubertal development, fertility, and hormone production; pathology can therefore produce acute surgical emergencies (e.g., torsion), chronic endocrine insufficiency, malignancy risk, sexually ambiguous genitalia, and long-term psychosocial and reproductive consequences. In recent years, major advances in molecular diagnostics, shifts toward multidisciplinary and patient-centred care, and evidence-based timing of interventions have redefined best practice for many conditions. This article synthesizes current concepts for diagnosis and management of the most common and consequential pediatric gonadal diseases, highlighting widely accepted guidelines and recent reviews.
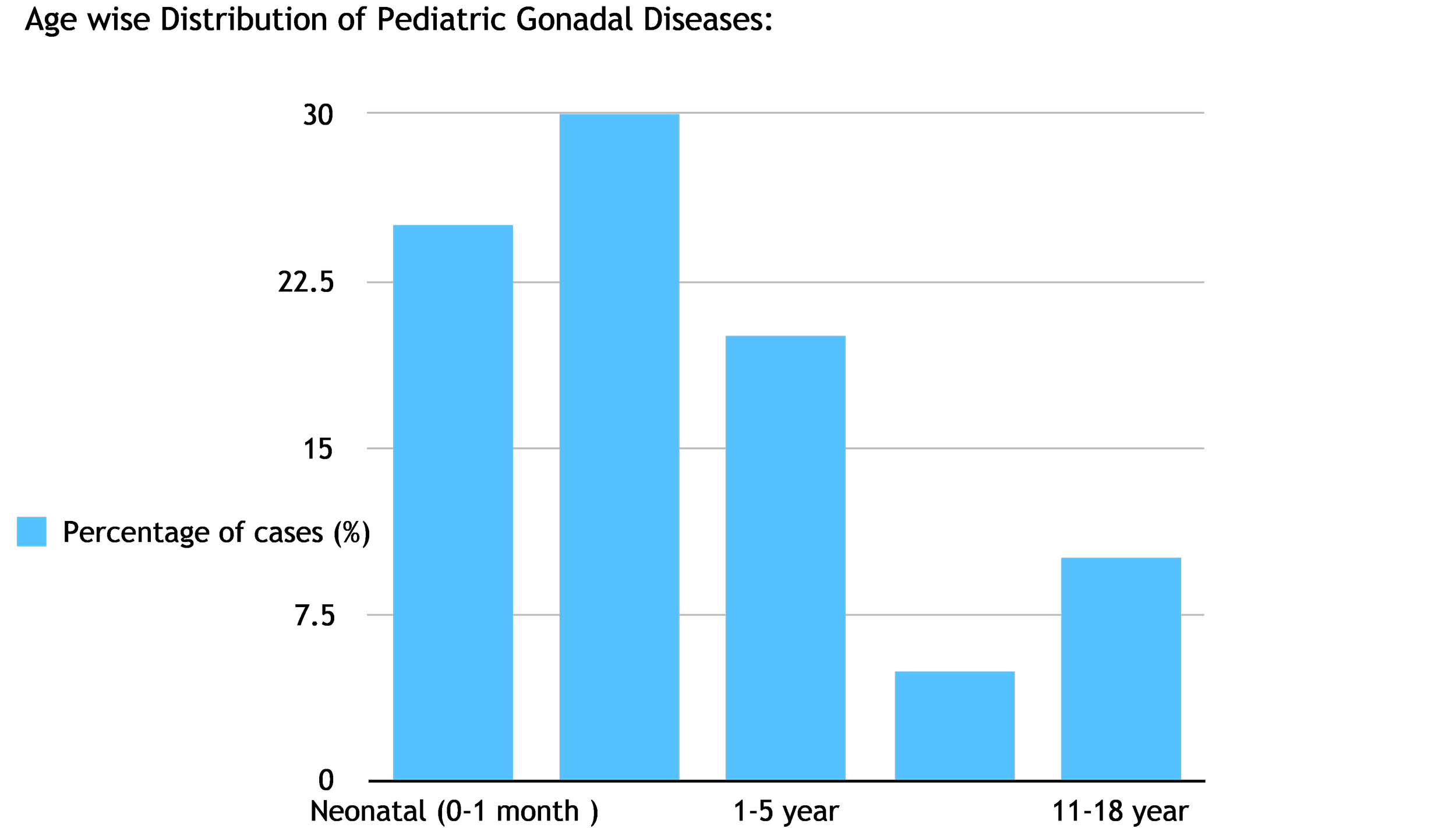
Epidemiology
Pediatric gonadal conditions are individually uncommon but cumulatively represent a significant burden due to potential long-term consequences. Examples:
Cryptorchidism (undescended testis): observed in up to 3–5% of full-term male neonates and higher in preterm infants; it is one of the most common congenital abnormalities requiring pediatric surgical care. Timely orchidopexy reduces malignancy and infertility risks.
Disorders/Differences of Sex Development (DSD): estimated incidence is between ~1:4,500 to 1:5,000 for the classical DSDs that present with genital ambiguity; broader variations in sex development may be more common. Management requires multidisciplinary care.
Pediatric gonadal tumours (germ cell tumours and sex-cord stromal tumours): relatively rare but account for a measurable proportion of pediatric solid tumours; prognosis has improved with combined surgical and platinum-based chemotherapy regimens.
Testicular torsion: an acute surgical emergency with an incidence of roughly 3.8 per 100,000 males younger than 18 years in some series; prompt diagnosis and detorsion are critical to salvage. (See surgical references in section below.)
Because incidence, age of presentation, and outcomes vary by specific disorder, clinicians should have a high index of suspicion and clear referral pathways to specialized centers.
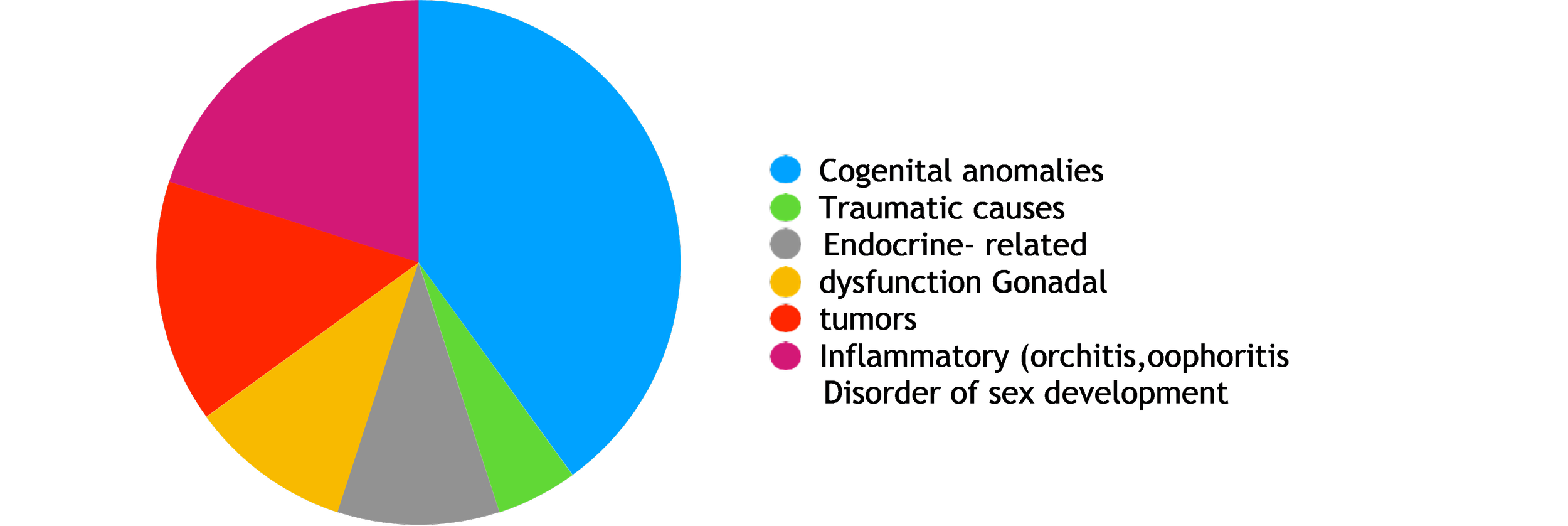
Classification and pathophysiology
For clinical use, pediatric gonadal conditions can be grouped into the following categories:
Developmental/Genetic disorders of gonadal formation and sex differentiation
46,XY gonadal dysgenesis (Swyer), 46,XX testicular DSD, mixed gonadal dysgenesis, congenital adrenal hyperplasia (46,XX with virilization), androgen insensitivity syndromes, enzyme defects, SRY translocations.
Anatomic anomalies and descent abnormalities
Cryptorchidism (undescended testis), ectopic testis, vanishing testis.
Acute surgical conditions
Testicular torsion, incarcerated inguinal hernia involving gonad, traumatic rupture. (Surgical literature: guidelines emphasize rapid triage.)
Neoplastic disease
Gonadal germ cell tumours (yolk sac tumour, teratoma, embryonal carcinoma, mixed GCT), sex cord-stromal tumours (Sertoli/Leydig cell tumours), gonadoblastoma in dysgenetic gonads.
Endocrine and autoimmune gonadal failure
Premature ovarian insufficiency (including autoimmune ovarian failure), primary testicular failure (e.g., anorchia, Klinefelter syndrome presentations in adolescence), autoimmune orchitis.
Infectious/traumatic causes
Post-infectious orchitis, gonadal injury.
Understanding the embryology of sex differentiation (timing of SRY expression, androgenic effects on internal/external genital development) aids targeted biochemical and genetic testing and informs counselling for sex assignment in infants with ambiguous genitalia. Contemporary consensus emphasizes family-centred multidisciplinary evaluation rather than immediate irreversible surgical decisions in neonates with ambiguous genitalia.
Clinical presentation and initial triage
Presentation varies by condition and age:
Neonate with ambiguous genitalia or atypical genital appearance: prioritized early evaluation in the first hours to days (electrolytes if salt-wasting CAH is possible), karyotype, pelvic ultrasound, endocrine tests. Shared decision-making and involvement of DSD multidisciplinary team (endocrinology, genetics, surgery/urology, psychology, neonatology) are recommended.
Undescended testis: detected on newborn exam, well-child visits; non-palpable vs. palpable distinction guides imaging and referral. Guidelines recommend early referral for surgical orchidopexy (timing discussed below).
Acute scrotum (torsion): sudden severe pain, swelling — urgent urologic/surgical evaluation and expedited detorsion; timelines for salvage are short (hours). (See surgical guidance.)
Mass or swelling: painless testicular mass or adnexal mass in female child/adolescent — evaluate for tumour markers (AFP, β-hCG, LDH), ultrasound, and early oncology referral.
Delayed or abnormal puberty: may indicate gonadal failure, endocrine axis disorders, or tumour- related hormone production.
Initial triage should always include stabilization (if acutely ill), assessment for life- or limb- threatening situations (salt wasting, torsion), and simultaneous initiation of baseline diagnostic tests when feasible.
Diagnostic approach
A scalable, pragmatic diagnostic algorithm:
Stabilize the patient (if necessary): address dehydration, electrolyte abnormalities (e.g., salt- wasting CAH in neonates).
Focused clinical exam and history: family history of DSD, infertility, consanguinity, maternal exposures, previous surgeries.
Laboratory tests
Neonatal ambiguous genitalia: serum electrolytes, 17-hydroxyprogesterone, cortisol, glucose, plasma renin activity (if concerned about salt wasting), karyotype or rapid QF PCR, baseline LH/ FSH/testosterone/AMH as appropriate.
Suspected gonadal tumour: serum AFP, β-hCG, LDH; CBC and basic metabolic panel.
Imaging:
Scrotal and pelvic ultrasound is first-line for gonadal location, masses, and evaluation of Mullerian structures in DSD. MRI can be used when ultrasound is non-contributory.
Genetic testing:
Karyotyping is essential in ambiguous genitalia. Chromosomal microarray and targeted gene panels or exome sequencing increasingly help identify etiologies (e.g., AR gene mutations, steroidogenic enzyme mutations, SRY translocations). Contemporary care increasingly uses next- generation sequencing panels in parallel with endocrine testing.
Specialized procedures:
Diagnostic laparoscopy for nonpalpable testis; surgical biopsy considered selectively in suspicious gonadal masses or dysgenetic gonads at risk for gonadoblastoma.
Importantly, many consensus statements emphasize multidisciplinary coordination so that investigations, invasive procedures, and counselling are performed in a considered manner.
Disorders/Differences of Sex Development (DSD)
Overview & classification. DSDs include conditions in which chromosomal, gonadal, or anatomic sex development is atypical. Classification by karyotype (46,XX; 46,XY; sex chromosome DSD) remains clinically useful. Common causes include congenital adrenal hyperplasia (CAH), androgen insensitivity syndrome (AIS), and gonadal dysgenesis.
Acute neonatal priorities. Rule out life-threatening CAH with serum electrolytes and 17- hydroxyprogesterone; start glucocorticoid and mineralocorticoid therapy when salt-wasting adrenal insufficiency is suspected. Rapid involvement of DSD team is advised.
Diagnostic testing. Karyotype, hormonal studies (basal and stimulation tests when indicated), pelvic/scrotal ultrasound, and targeted genetic testing. AMH and anti-Müllerian hormone can help determine presence of testicular tissue.
Management principles
Multidisciplinary, individualized, with prioritization of: Immediate medical stabilization when required.
Establishing a likely etiologic diagnosis before irreversible interventions.
Engaging family in shared decision-making, with support from mental health professionals and, when appropriate, advocacy groups. International consensus advises against early cosmetic genital surgery without informed, multidisciplinary consultation except in strict, justified circumstances.
Long-term care. Lifelong endocrine follow-up, fertility counselling (fertility may be preserved or impaired depending on diagnosis), and psychosocial support.
Cryptorchidism (Undescended Testis)
Epidemiology & rationale for repair. Cryptorchidism is common in neonates, with higher prevalence in preterm infants. Undescended testis is associated with reduced fertility potential and increased risk of germ cell tumours if not corrected. Contemporary guidelines recommend early surgical correction to optimize outcomes.
Timing of orchidopexy. Most recent pediatric urology guidelines recommend orchidopexy between 6 and 12 months of age (some guidelines allow up to 18 months) because spontaneous descent beyond 6 months is unlikely and early correction may improve fertility potential.
Workup. Careful physical exam (palpable vs. nonpalpable testis). Imaging is not routinely recommended for palpable testis but may be used selectively. Laparoscopy is the diagnostic and therapeutic approach for nonpalpable testes. Hormonal therapy (hCG or GnRH) has limited and inconsistent efficacy and is not recommended as routine first-line therapy for undescended testis.
Follow-up. Postoperative follow-up to assess testicular size and position, and long-term surveillance for fertility and malignancy risks when clinically indicated.
Testicular Torsion
Clinical urgency. Torsion causes ischemia; testicular salvage depends on prompt detorsion within hours. Clinical diagnosis supported by Doppler ultrasound; however, if clinical suspicion is very high, do not delay surgery for imaging. Contralateral orchidopexy is generally performed at time of surgery. (Refer to surgical guidelines.)
Management. Immediate urologic consultation and operative exploration with detorsion and bilateral fixation where possible. Outcomes are time-dependent.
Gonadal Tumours (Pediatric Gonadal Neoplasms)
Epidemiology & pathology. Gonadal tumours in children are largely germ cell tumours (GCTs) in both sexes; age distribution and behaviour differ from adult tumours. The 2022 WHO updates and multiple pediatric oncology reviews emphasize risk-adapted treatment strategies.
Diagnosis. Testicular or ovarian mass on imaging, plus tumour markers (AFP, β-hCG, LDH) guide initial staging and pathologic suspicion. Surgical excision is diagnostic and often therapeutic; staging and histologic subtype determine adjuvant therapy.
Management
Mature teratoma (benign in children): surgical excision is often curative.
Malignant GCTs (yolk sac tumour, embryonal carcinoma, choriocarcinoma) typically require platinum-containing chemotherapy in addition to surgery for advanced stages.
Gonadoblastoma risk in dysgenetic gonads: prophylactic gonadectomy is considered in dysgenetic gonads with Y-chromosome material because of high malignant transformation risk. Multidisciplinary case discussion is essential.
Outcomes. Improved substantially with modern chemotherapy; relapse risk depends on stage and histology. Early multidisciplinary care (pediatric surgery, oncology, pathology) is key.
Autoimmune and Genetic Gonadal Failure
Autoimmune ovarian insufficiency and primary testicular failure are less common in children but have profound implications for growth, puberty, and fertility. Evaluation includes gonadotropins (LH/FSH), sex steroid levels, and autoimmune serologies where appropriate. Management includes hormone replacement, fertility counselling, and consideration of fertility preservation in adolescents. Evidence base for interventions in prepubertal children is limited; individualized planning is necessary.
Fertility preservation and ethical considerations
Fertility preservation is an emerging priority in pediatric gonadal disorders and oncology. In prepubertal children, options are limited (sperm cryopreservation not feasible before spermatogenesis), but experimental options such as testicular tissue cryopreservation exist in specialized centers. For post-pubertal adolescents, standard sperm or oocyte preservation strategies apply. The decision to pursue fertility preservation must balance the natural history of the disease, urgency of treatment, ethical considerations, and family preferences. Multidisciplinary counseling with reproductive specialists is recommended early in the care pathway.
DSD management introduces ethical complexity regarding irreversible surgeries in infancy; major consensus statements endorse family-centred shared decision-making and restraint regarding early cosmetic genital surgery unless medically indicated.
Long-term follow-up and transition to adult care
Many pediatric gonadal conditions (e.g., DSD, gonadal dysgenesis, cryptorchidism repaired in infancy, survivors of gonadal tumours) require life-long surveillance for endocrine health, sexual function, fertility, and psychosocial wellbeing. Establishing structured transition pathways to adult endocrinology/urogenital oncology services improves continuity of care and long-term outcomes. Registries and standardized follow-up protocols are encouraged to improve evidence for long- term risks.
Table 1. Differential diagnosis for a neonate with ambiguous genitalia

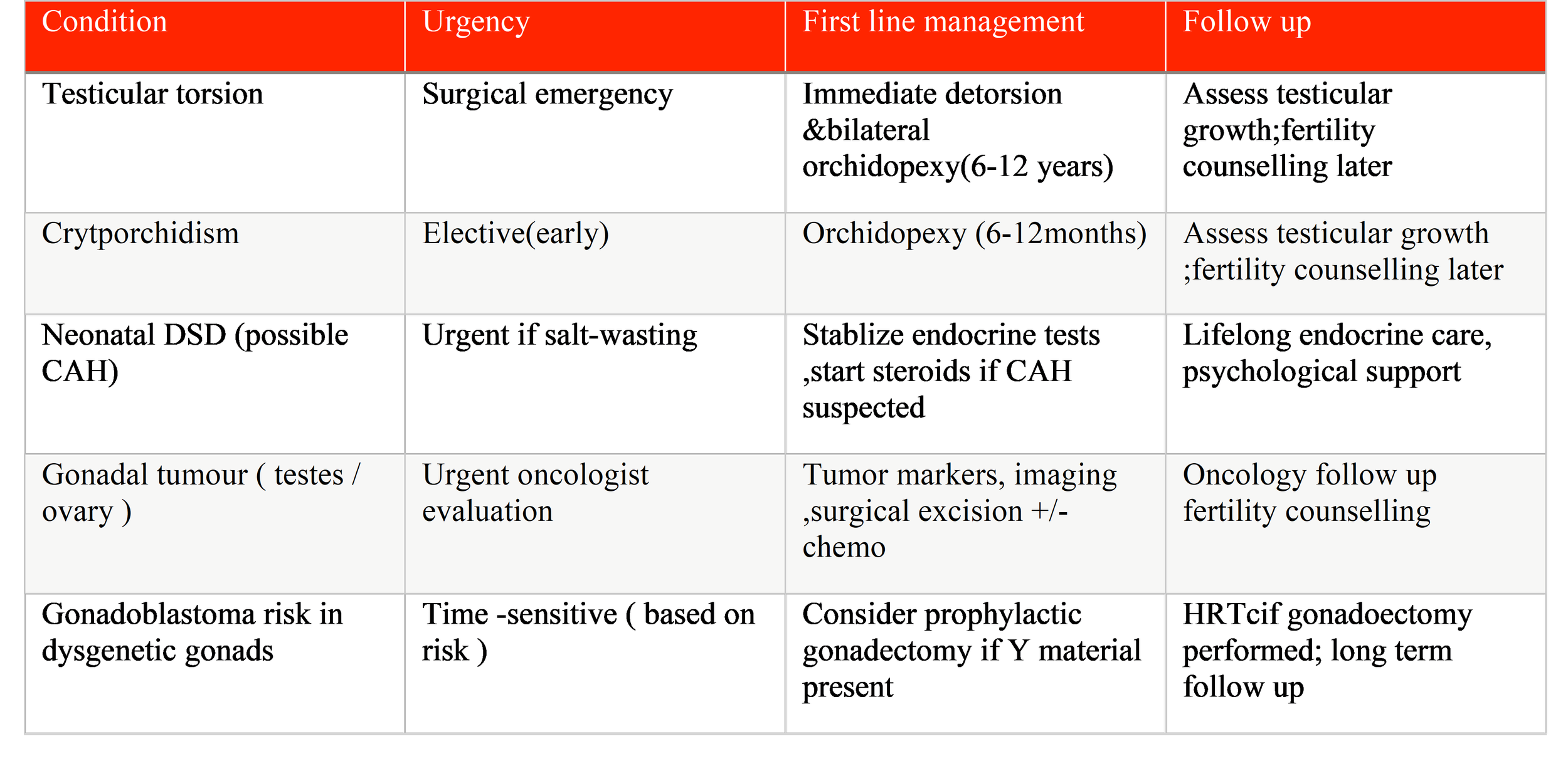
Diagnostic & management algorithms
Algorithm: Neonate with ambiguous genitalia (summary)
Rapid assessment: ABCs, check electrolytes (spot check), consider CAH as potential emergency.
Order: karyotype, serum 17-OH progesterone, cortisol, glucose, AMH, pelvic ultrasound.
Convene DSD MDT (endocrinology, genetics, surgery/urology, psychology).
Discuss provisional diagnosis and family-centred plan; avoid irreversible surgeries until appropriate evaluation and counselling.
Algorithm: Boy with nonpalpable testis
Confirm on exam; repeat in warm room and with age-appropriate techniques.
If nonpalpable: refer to pediatric urology; diagnostic laparoscopy often indicated.
Plan orchidopexy or orchiectomy as indicated based on intraoperative findings.
Recent evidence highlights and guideline recommendations
Consensus statements and guidelines emphasize multidisciplinary care, slow and careful consideration before irreversible genital surgery, and early stabilization for CAH in neonates. These remain central tenets of modern care for DSD.
For cryptorchidism, high-quality guideline updates support orchidopexy between 6–12 months of age to optimize future fertility potential and reduce malignant transformation risk.
Pediatric germ cell tumours management has evolved with organ-sparing approaches for low-risk gonadal lesions and risk-adapted chemotherapy regimens for malignant subtypes; contemporary WHO classification and pediatric oncology reviews summarize these advances.
Research gaps and future directions
Long-term outcomes and registries. More registry-based, longitudinal studies are needed to quantify fertility, sexual function, malignancy risk, and psychosocial outcomes for diverse pediatric gonadal conditions.
Fertility preservation in prepubertal children. Experimental techniques require standardized protocols and ethical frameworks.
Genotype–phenotype correlations. Widespread access to next-generation sequencing and centralization of genotype–phenotype databases would improve diagnostic precision and personalized treatment.
Psychosocial outcomes and decision-making. Comparative research on different care and counselling models is necessary to optimize shared decision-making for families of children with DSD.
Practical recommendations
Prioritize stabilization for neonates with suspected CAH and immediate surgical care for suspected torsion.
Use a multidisciplinary team for DSD evaluation and management; avoid irreversible procedures without full evaluation and shared decision processes.
Refer cryptorchidism for surgical correction early (target 6–12 months).
Use tumour markers (AFP, β-hCG) and ultrasound for suspected gonadal tumours; treat with surgery ± chemotherapy depending on histology and stage.
Plan for transition to adult care and consider fertility preservation discussions opportunely.
Conclusion
Pediatric gonadal diseases represent a diverse and complex group of conditions with implications that extend far beyond childhood, affecting sexual development, endocrine function, fertility potential, oncologic risk, and psychosocial well-being. Advances in developmental biology, molecular genetics, imaging modalities, and pediatric surgical and oncologic care have significantly refined the diagnostic and therapeutic approaches to these disorders. Contemporary management emphasizes early and accurate diagnosis, timely intervention for urgent and potentially reversible conditions such as congenital adrenal hyperplasia and testicular torsion, and evidence-based timing of elective procedures such as orchidopexy to optimize long-term outcomes.
A central theme in current practice is the shift toward multidisciplinary, patient- and family- centered care, particularly in disorders/differences of sex development, where ethical considerations, shared decision-making, and long-term psychosocial support are integral to management. Improved understanding of genotype–phenotype correlations and increasing availability of advanced genetic testing have enhanced diagnostic precision and allowed for more individualized treatment strategies. In parallel, risk-adapted approaches to pediatric gonadal tumors and growing attention to fertility preservation have improved survival and quality of life for affected children and adolescents.
Despite these advances, significant gaps remain in long-term outcome data, fertility preservation options for prepubertal patients, and standardized transition-of-care models into adulthood.
Future research should focus on longitudinal registries, integration of genomic medicine into routine practice, and development of evidence-based psychosocial and ethical frameworks. Strengthening multidisciplinary collaboration and ensuring lifelong follow-up will be essential to translating current concepts into sustained improvements in health and quality of life for patients with pediatric gonadal diseases.
References
1. Cools M, Nordenström A, Robeva R, Hall J, Westerveld P, Flück C, Köhler B, Berra M, Springer A, Schweizer K, et al. Caring for Individuals with a Difference of Sex Development (DSD): A Consensus Statement. Nat Rev Endocrinol. 2018.
2. Guerrero-Fernández J, et al. Management guidelines for disorders/different sex development (DSD). Anales de Pediatría (English Edition). 2018.
3. Bode PK, et al. Germ cell tumors in children. Review (PMC). 2025. (Discusses WHO classification updates and treatment.)
4. Kolon TF, et al. Evaluation and Treatment of Cryptorchidism (AUA/paediatric guideline resources). 2025 guideline summary.
5. Weil BR, et al. Management of Germ Cell Tumors in Pediatric Patients. MAGIC consortium review. 2021.
6. Gavrilovici C, et al. Orchidopexy Timing and Follow Up: From Guidelines to... 2025 review (supports early orchidopexy timing).
7. Suorsa-Johnson K, et al. Shared decision making in pediatric differences... (Editorial on care models) 2023.